- 카테고리 Transcriptomics > RNA-seq-transcriptomics
- 수정일2025-10-31 18:37:00
- 레퍼런스
RNA-seq Analysis Pipeline은 RNA-Seq 데이터를 처리하고 유전자 발현에 대한 통계적 분석을 수행하는 것을 목표로 합니다. 이 파이프라인은 유전자의 발현 수준을 이해하고 해석하기 위해 실험 데이터의 품질을 평가하고 정제하며, 정렬, 발현 수준 계산, 통계적 분석, 결과 시각화로 구성되어 있습니다.파이프라인의 초기 단계에서는 실험 데이터의 품질 평가와 정제가 이루어집니다. FastQC를 사용하여 실험 데이터의 품질을 검사하고 평가하며, Cutadapt를 활용하여 시퀀싱 어댑터 및 낮은 품질의 리드를 효과적으로 정제합니다.다음으로, STAR 2를 이용하여 리드를 정확하게 유전체에 정렬하고, 각 유전자의 발현 위치를 정밀하게 파악합니다. 그 후, Rsubread 라이브러리의 FeatureCounts를 활용하여 정렬된 리드를 각 유전자에 할당하여 발현 수준을 정량화하고 Count Matrix를 생성합니다.이어지는 단계에서는 R 기반의 edgeR과 limma를 사용하여 발현 수준의 통계적 차이를 식별하고 각 유전자의 발현 변동을 분석합니다. 이 과정에서 control과 test 샘플에는 각각 최소 두 개 이상의 생물학적 복제 샘플이 포함되어야 통계적 분석이 가능하다는 점에 유의해야 합니다. 복제가 없는 경우 잔차 유도가 0이 되어 분석이 실패하거나 결과가 신뢰성을 잃을 수 있습니다. 또한, R 기반의 fgsea를 활용하여 gene set 간의 풍부도를 평가하고, 다양한 시각화 도구를 활용하여 효과적으로 표현합니다. 마지막으로, fgsea의 결과 파일을 이용하여 여러 R 패키지를 통해 데이터 시각화, 그래픽 생성 등 실험 결과를 자세히 분석하고 시각화합니다.전체적으로, 최상위 입력 데이터인 fastq 형식의 RNA-seq raw data로부터 시작하여 품질 보고서인 fastqc.report.html을 생성하고, MA plot, correlation plot, network, volcano plot, heatmap 등의 다양한 시각화 자료를 통해 유전자 발현 및 풍부도를 시각적으로 확인할 수 있습니다.
Bio-Express RNA-seq Alternative-splicing Pipeline (이하 AS)은 유전자 발현의 전사체 수준에서 크게 5가지 type의 splicing 양상을 확인할 수 있다.Alternative splicing은 하나의 유전자를 구성하는 복수의 exon (coding region)간의 조합에 따라 여러 transcripts (isoforms)가 생성되며, 이에 따라 하나의 유전자라도 서로 다른 구조를 갖는 단백질이 만들어짐에 따라 기능이 다른 유전자로써 역할을 하게된다. 이러한 메커니즘을 통해 단백질의 폭넓은 capacity 를 확보할 수 있으며, 다양한 분자적 역할이 가능하다.

(출처: From Wikipedia, the free encyclopedia)
기본적인 5가지 형태의 이벤트는 아래와 같다.
1. SE (Exon skipping | cassette exon)
2. MXE (Mutually exclusive exons)
3. A5SS (Alternative donor site)
4. A3SS (Alternative acceptor site)
5. RI (Intron retention)
.png)
AS 분석 파이프라인은 아래와 같은 흐름으로 진행됨.
1. Quality Control, 시퀀싱 품질 관리 (by FastQC)
2. Trimming, 아답터 및 low quality 제거 (by Cutadapt, Trimmomatic)
3. Mapping, 레퍼런스 alignment (by STAR, HISAT2)
4. AS detection, 선택적 스플라이싱 탐색 (by rMATs)
5. Visualization, AS 결과 시각화 (by ggsashimi)
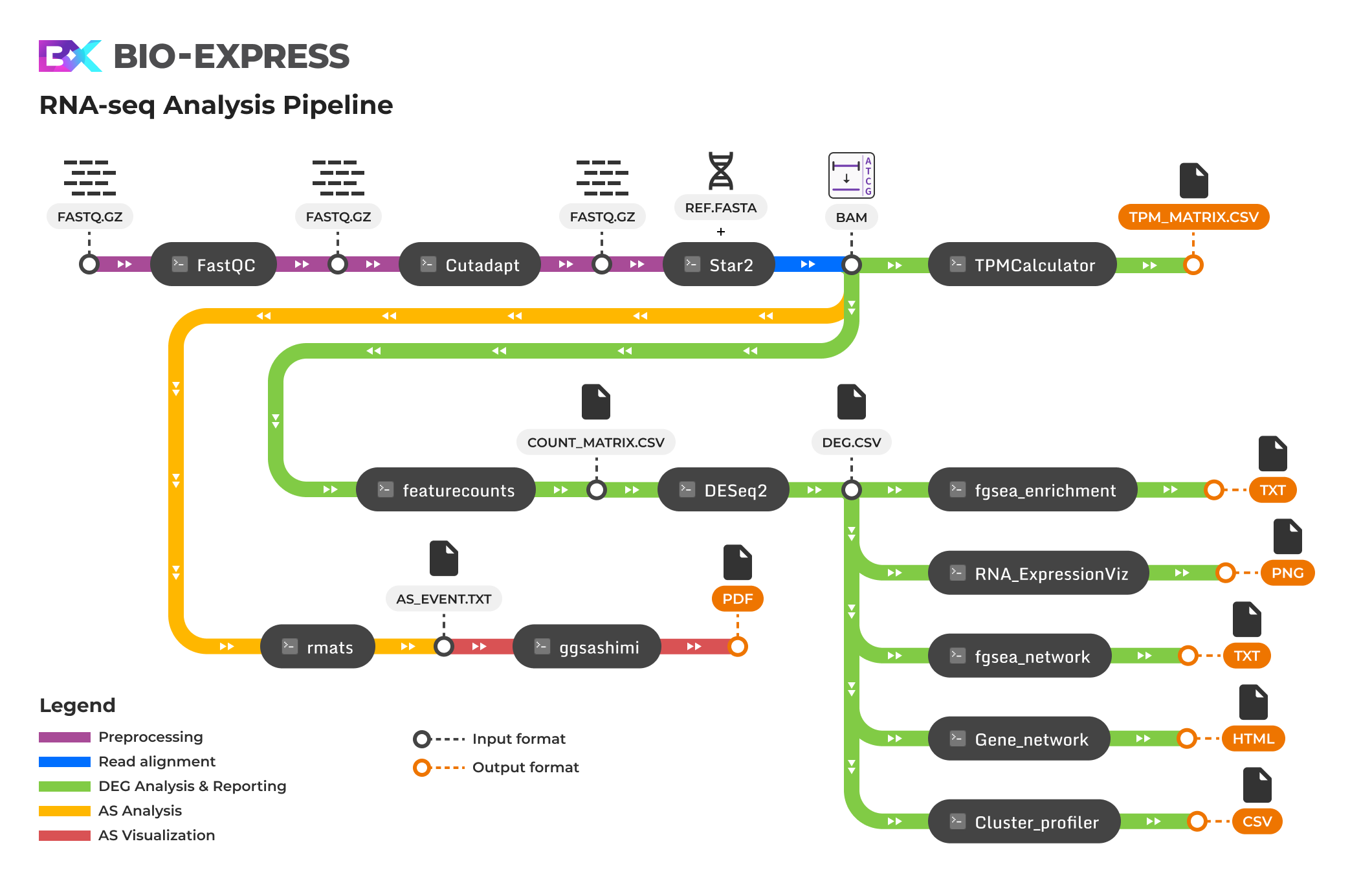
파이프라인 모듈
FastQC
FastQC는 고속 염기서열 분석(high throughput sequence) 데이터의 품질 관리를 위한 분석도구입니다. 이 프로그램은 FASTQ 형식의 서열 데이터를 읽어들여 여러 품질 관리(Qaulity Control) 검사를 수행하고 결과는 HTML 기반의 보고서로 출력합니다. FastQC는 전반적인 품질 문제에 대한 개요 정보를 제공하며, 쉽게 확인할 수 있는 요약된 그래프와 테이블을 포함합니다. FastQC는 FASTQ 형식의 파일이 입력 파일로 사용되며, 출력 결과는 리포트 html 파일과 zip 형식의 압축 파일이 생성됩니다.
주요사항
- FastQC는 자바 애플리케이션입니다. 실행하기 위해서는 시스템에 적절한 자바 실행 환경(Java Runtime Environment, JRE)이 설치되어 있어야 합니다. 따라서 FastQC를 실행하기 전에 먼저 적절한 JRE가 설치되어 있는지 확인해야 합니다. 다양한 종류의 JRE를 사용할 수 있지만, 저희가 테스트해본 것은 최신 오라클 런타임 환경과 adoptOpenJDK 프로젝트의 JRE입니다. 64비트 JRE를 다운로드하여 설치하고, 자바 애플리케이션이 시스템 경로(path)에 포함되도록 설정해야 합니다(대부분의 설치 프로그램이 이를 자동으로 처리해줍니다).
실행 명령어 예시
$program_dir/fastqc –t 6 –o $OUTPUT_DIR $INPUT_DIR/$READ
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o) |
결과
-
.png)
Basic Statistics 테이블은 주어진 FASTQ 파일에 대한 간단한 통계적 정보를 제공합니다. 일반적으로 다음과 같은 정보를 포함합니다. Filename : 분석된 파일의 이름 또는 경로 File type : FASTQ 파일의 종류 Encoding : 품질 점수 인코딩 방식 Total Sequence : 총 서열 수 Filtered Sequences : Read 품질이 좋지 않은 서열 수 Sequence length : 서열의 길이 %GC : 서열에서의 GC 백분율
-
.png)
X축은 리드의 염기 위치를 나타내며, Y축은 품질 점수를 의미합니다. 점수가 높을수록 품질이 좋습니다. 중앙의 빨간색 선은 중앙값을 나타내고, 노란색 박스는 사분위간 범위(25~75%)를 의미합니다. 위쪽 및 아래쪽의 위스커(whisker)는 각각 10% 및 90% 포인트를 나타냅니다. 파란색 선은 평균 품질을 의미합니다. 어떤 염기의 하위 사분위수가 10 미만이거나 중위값이 25보다 작을 경우 경고로 간주됩니다. 또한, 어떤 염기의 하위 사분위수가 5 미만이거나 중위값이 20보다 작으면 오류로 간주됩니다.
-
.png)
색을 이용하여 각 타일의 품질을 나타내며, 파란색은 품질이 높음을 나타내고 빨간색은 품질이 낮음을 나타냅니다. 각 타일의 품질을 모든 타일의 평균 품질과 비교하여 예상 패턴과의 편차를 식별 할 수 있습니다. 특정 타일의 품질이 지속적으로 좋지 않으면 물리적 결함이나 오염 등 셀의 특정 영역에 문제가 있음을 나타낼 수 있습니다. 이상적으로는 모든 타일이 높은 품질을 보여야 하면 플롯에서 더 차가운 색상으로 표시됩니다. 이 플롯은 Illumina 라이브러리에서만 나타납니다.
-
.png)
X축은 리드의 전체 길이에 대한 평균 품질 점수를 나타내고, Y축은 해당 품질 점수를 갖는 읽기의 수를 나타냅니다. 시퀀스의 시퀀싱 품질은 해당 시퀀스에 대한 정확성과 신뢰성을 나타내며, 품질 점수가 높을수록 오류 발생 가능성이 낮다는 것을 의미합니다. 만약 시퀀싱 실행이 전반적으로 낮은 품질을 보인다며, 시퀀싱 화학 문제나 샘플 준비 문제 등이 있을 수 있습니다. 품질 점수가 기록되지 않은 BAM/SAM 파일의 경우 확인할 수 없습니다. 품질 점수가 Phred 척도 기준 최고 품질 점수 27점(오류율 0.2%) 미만일 경우 경고 발생, 20점(오류율 1%) 미만일 경우 오류입니다.
-
.png)
X축은 리드의 포지션을 나타내고, Y축은 시퀀싱한 리드에서 각 base의 전체 비율을 나타냅니다. 좋은 품질의 시퀀싱 샘플에서는 각 위치의 염기 비율을 나타내는 4개의 선이 평행하고 서로 가까워야 합니다. 그러나 선이 일부 위치에서 엉키거나 얽히면 과도하게 표현된 시퀀스가 오염되었음을 나타낼 수 있습니다. 또한, A/T 또는 G/C 염기의 비율이 어떤 위치에서는 10% 이상 차이나면 경고 발생, 20%를 초과하면 오류입니다.
-
.png)
시퀀스에서 G와 C 뉴클레오티드의 백분율 비율을 나타냅니다. 이를 통해 DNA 또는 RNA 시퀀스의 특성을 이해할 수 있습니다. 시퀀스의 GC 함량은 DNA 안정성, 서열의 물리적 특성, 유전자 발현에 영향을 미칠 수 있으므로 중요한 지표 중 하나입니다. X축은 GC contents의 비율을 나타내고, Y축은 시퀀스의 총량을 나타냅니다. 정규 분포와 편차 합계가 전체 리드의 15%를 초과하면 경고, 30%를 초과하면 오류입니다.
-
.png)
시퀀싱 리드의 각 위치에서 발견된 N 비율을 나타내며 일반적으로 매우 낮습니다. 그러나 어떤 위치에서는 N 비율이 5%를 초과하면 시퀀싱 시스템에 문제가 있을 수 있다는 경고로 간주되며, 20%를 초과하면 오류로 간주됩니다. 데이터의 품질이 높은지 확인하고 시퀀싱 읽기의 정확성에 영향을 줄 수 있는 문제를 식별하려면 시퀀싱 중에 N 비율을 모니터링하는 것이 중요합니다. N 비율이 권장 임계값을 초과하는 경우 문제의 심층적인 분석이 필요할 수 있습니다.
-
.png)
시퀀싱 데이터에서 각 리드의 길이에 대한 분포를 나타냅니다. 이 그래프의 X 축은 시퀀스 길이를, Y 축은 리드 수를 나타냅니다. 시퀀싱 데이터에서 발견된 리드의 길이가 어떻게 분포되어 있는지를 시각적으로 확인할 수 있습니다. 이 그래프를 통해 시퀀싱 데이터 세트의 리드 길이 분포를 파악할 수 있으며, 예상치 못한 리드 길이나 이상한 분포를 감지하여 데이터의 품질을 평가하는 데 도움이 됩니다. 일반적으로 시퀀스 길이 분포는 일정하거나 특정한 패턴을 따르지만, 비정상적인 분포는 시퀀싱 데이터에 문제가 있을 수 있다는 신호일 수 있습니다.
-
.png)
중복된 시퀀스가 전체의 20% 이상일 경우 경고, 50% 이상일 경우 오류입니다.
-
.png)
시퀀싱 데이터에서 빈번하게 등장하는 시퀀스를 나타내는 테이블입니다. 이 테이블은 시퀀싱 데이터에서 특정 시퀀스가 기대보다 더 자주 나타나는 경우를 식별합니다. 실험과정에서 발생한 오류, PCR 이중성, 어댑터 오류 또는 샘플의 비정상적인 특정으로 인해 발생할 수 있습니다. 해당 내용을 확인하고 잠재적인 문제를 식별하는 것은 시퀀싱 데이터의 정확성과 신뢰성을 높이는 데 도움이 됩니다.
-
.png)
시퀀싱 데이터에서 발견된 어댑터 시퀀스의 누적 백분율을 보여주는 차트입니다. 시퀀스가 발견된 위치에 따라 백분율이 증가하며, 시퀀스가 리드의 끝까지 존재하는 동안 계산됩니다. 어댑터의 비율을 확인하여 데이터의 품질을 평가하며, 어댑터 시퀀스가 발견되는 비율이 높을수록 시퀀싱 데이터에 오류가 포함될 가능성이 높아지므로, 5%를 초과하면 경고, 10%를 초과하면 오류입니다.